Publications
Research publications from Jo Lab
DuAL-Net: A Dual-Network Approach for Alzheimer's Disease Risk Prediction Using APOE-Centered Regional WGS Data
This study presents DuAL-Net (Dual Approach Local-global Network), a hybrid framework that integrates local genomic patterns with global biological knowledge for Alzheimer's disease risk prediction...
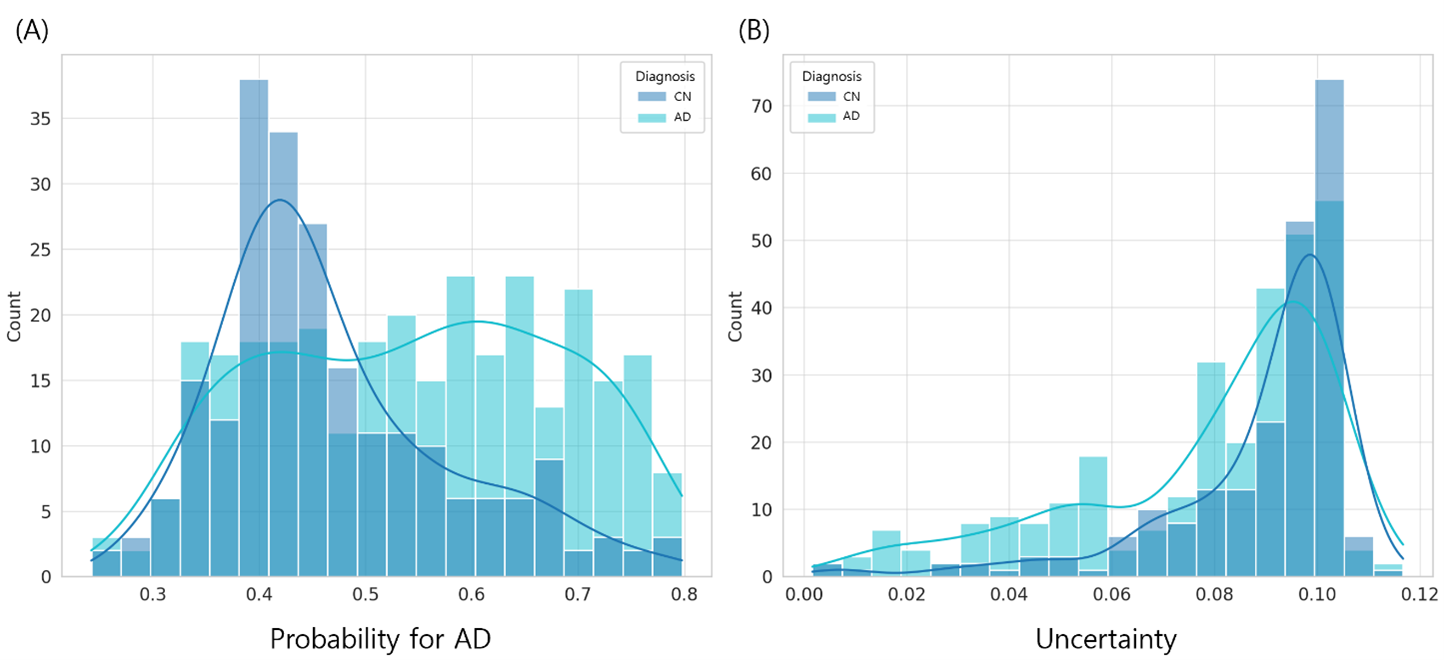
Uncertainty-aware genomic classification of Alzheimer's disease: a transformer-based ensemble approach with Monte Carlo dropout
TrUE-Net combines transformer and random forest models with Monte Carlo Dropout to provide uncertainty-aware AD classification from WGS data. Analyzing 1,050 individuals (607 AD, 443 controls) from...
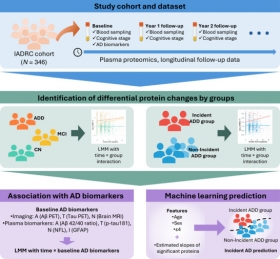
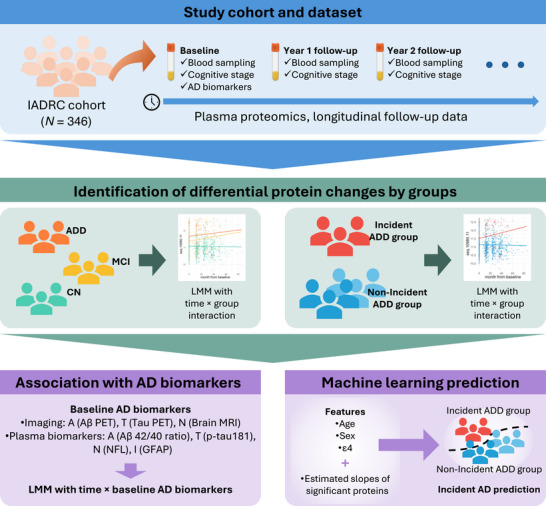
Longitudinal plasma proteomics: relation to incident Alzheimer's disease dementia and biomarkers
Longitudinal proteomics analysis identified dynamic changes in plasma proteins associated with AD progression. Seven proteins (ACES, C7, ZCD1, IL-17C, CC055, SO5A1, IGFALS) showed significant assoc...
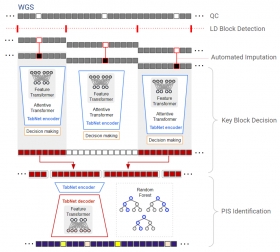
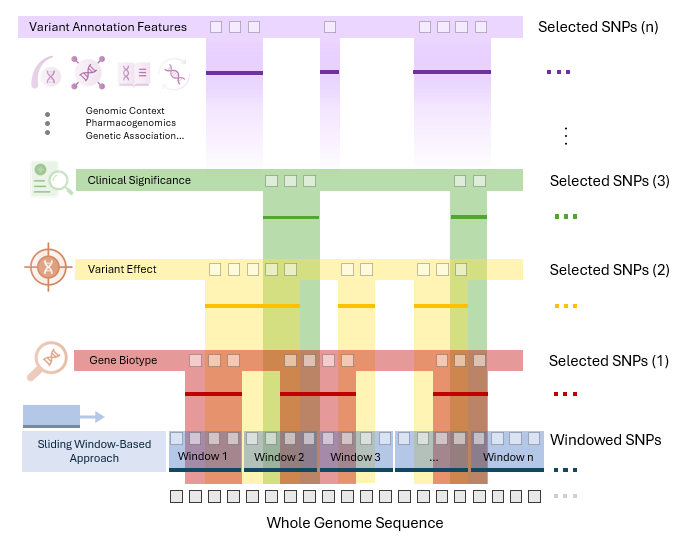
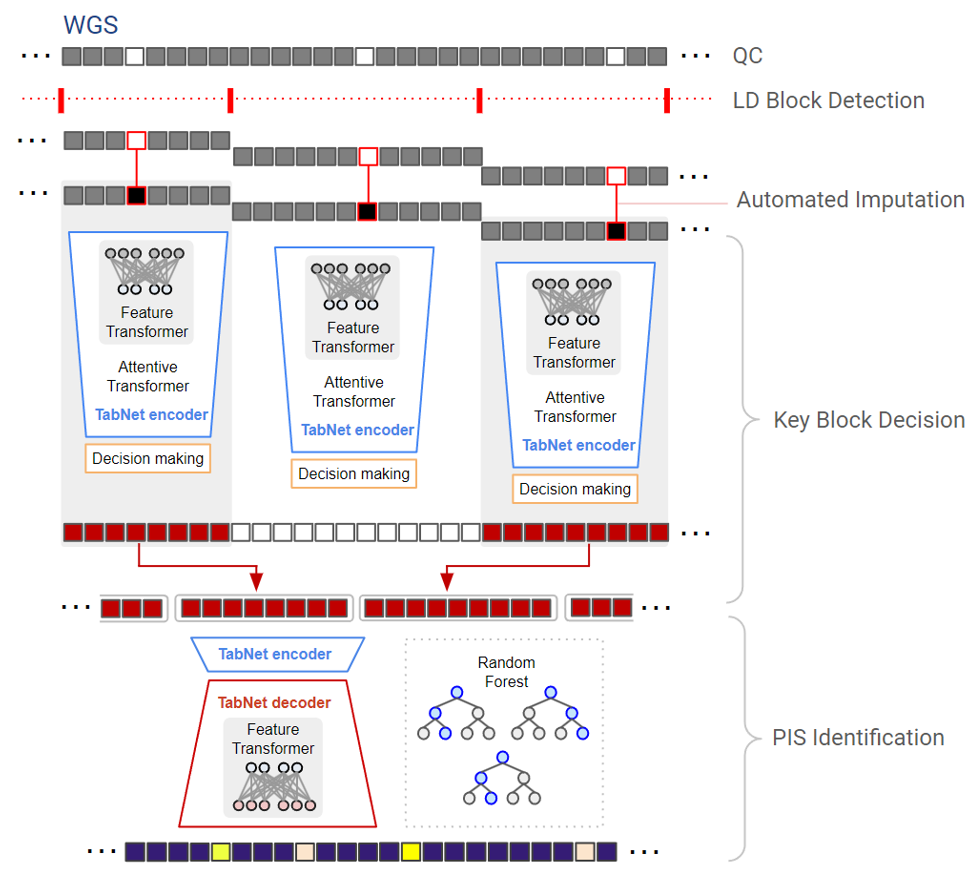
LD‐informed deep learning for Alzheimer's gene loci detection using WGS data
Deep‐Block is a multi‐stage deep learning framework designed to detect AD associated genetic loci in large‐scale WGS data. It segments the genome based on linkage disequilibrium, applies sparse att...
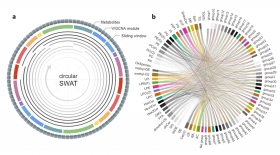
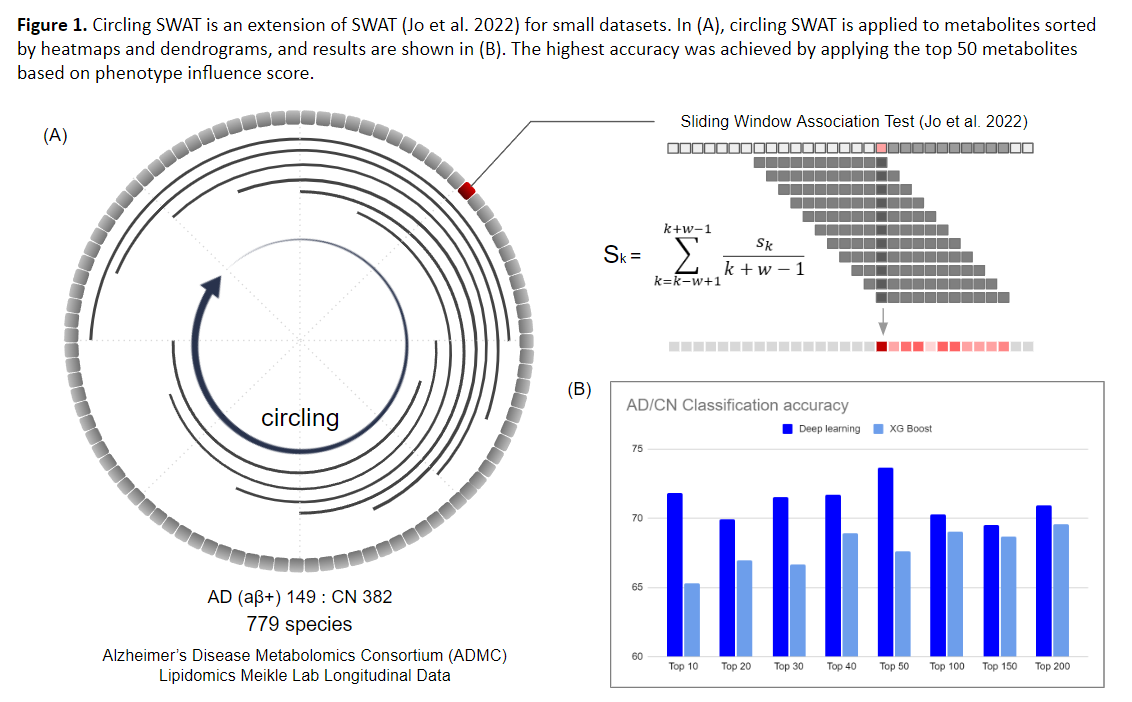
Circular-SWAT for deep learning based diagnostic classification of Alzheimer’s disease: Application to metabolome data
This study introduces the Circular-Sliding Window Association Test (c-SWAT), a methodology designed to enhance the diagnostic classification of AD using serum-based metabolomics data, with a focus ...
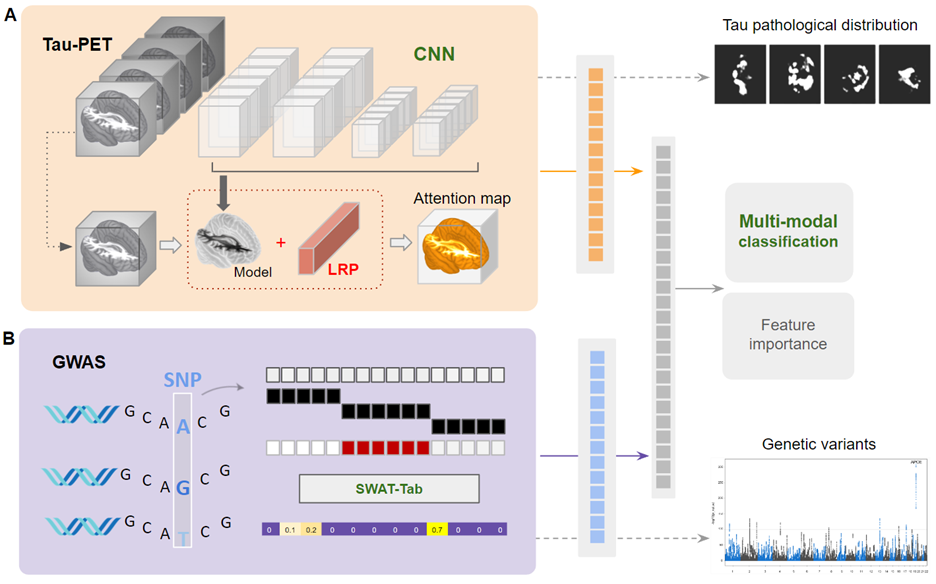
Deep Learning-based Integration of Neuroimaging and Genetic Data for Classification of Alzheimer's Disease
This study introduces a new deep learning method using CNNs to analyze tau PET images and identify Alzheimer's Disease (AD) related patterns. The method achieved a 90.8% accuracy in classifying AD ...
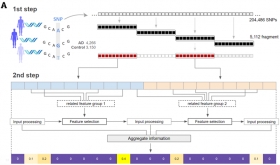
Deep Learning-based SWAT-Tab Approach for Identifying Genetic Variants using Whole Genome Sequencing
The study introduces SWAT-TAB, an evolved form of SWAT-CNN, optimized for identifying genetic variants in Alzheimer's disease (AD). It utilizes the Tabnet algorithm to meticulously select relevant ...
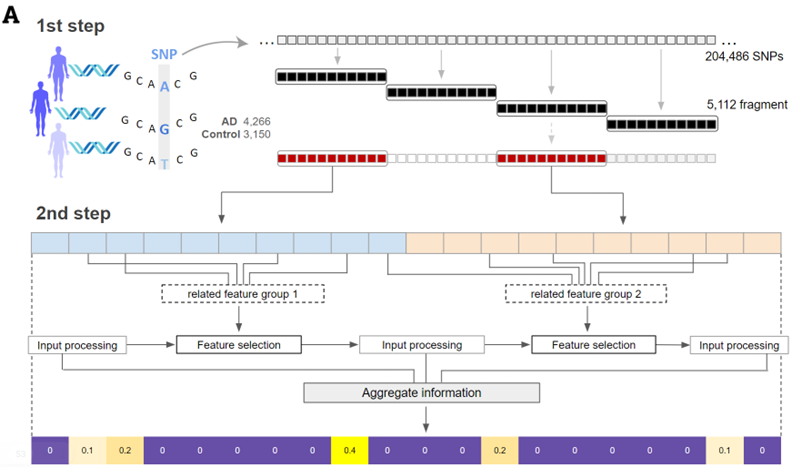
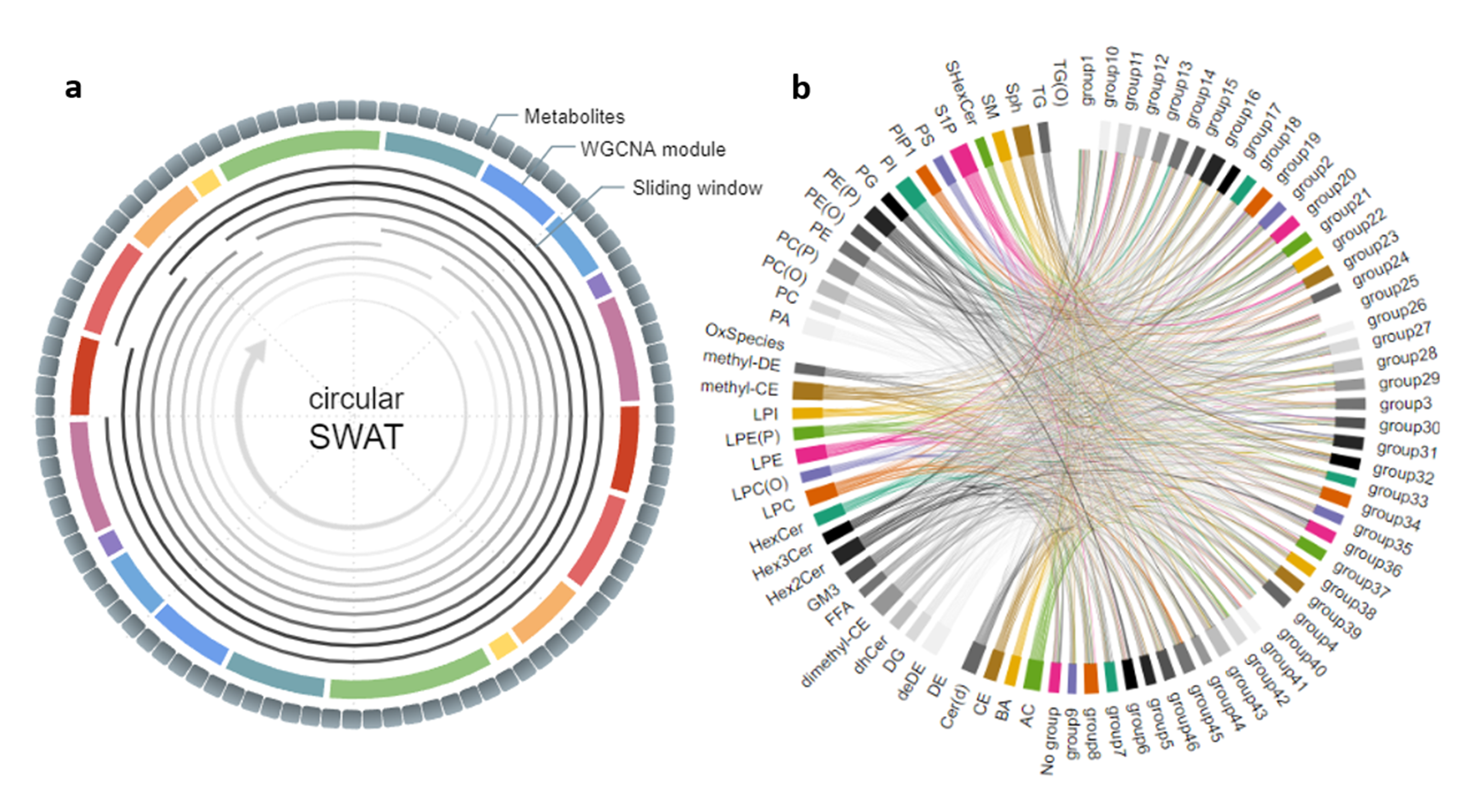
Novel circling SWAT for deep learning based diagnostic classification of Alzheimer’s disease: Application to metabolome data
We used serum-based cross-sectional lipidome data with 781 lipids from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) including 216 cognitively normal (CN), 635 MCI, and 382 dementia (AD). ...
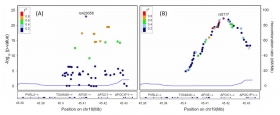
Deep learning-based identification of genetic variants: application to Alzheimer’s disease classification
We propose a novel three-step approach (SWAT-CNN) for identification of genetic variants using deep learning to identify phenotype-related single nucleotide polymorphisms (SNPs) that can be applied...
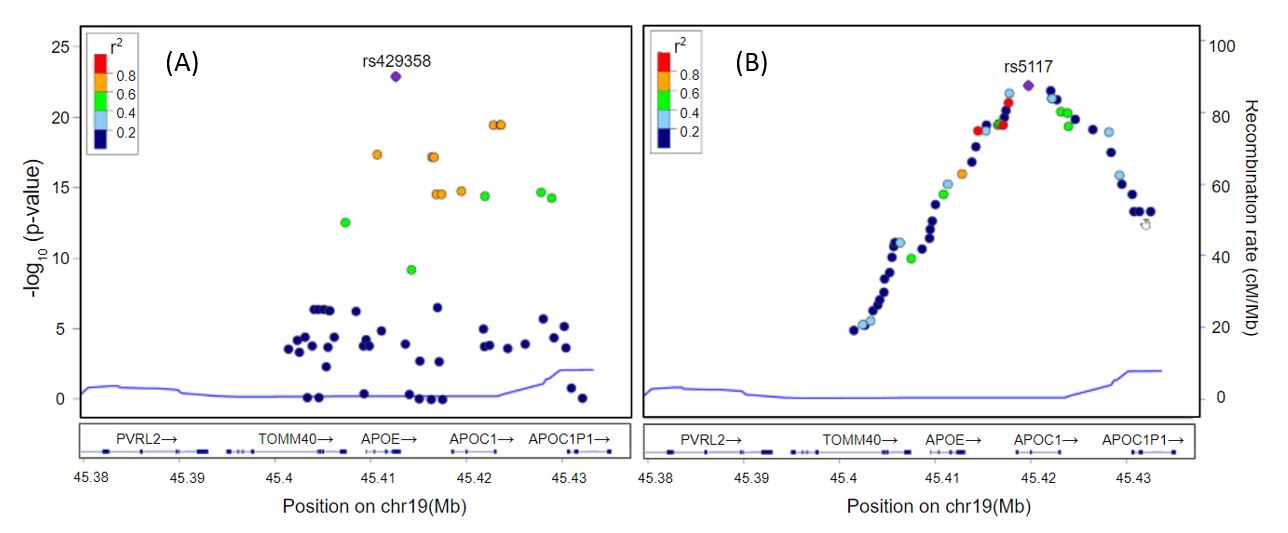
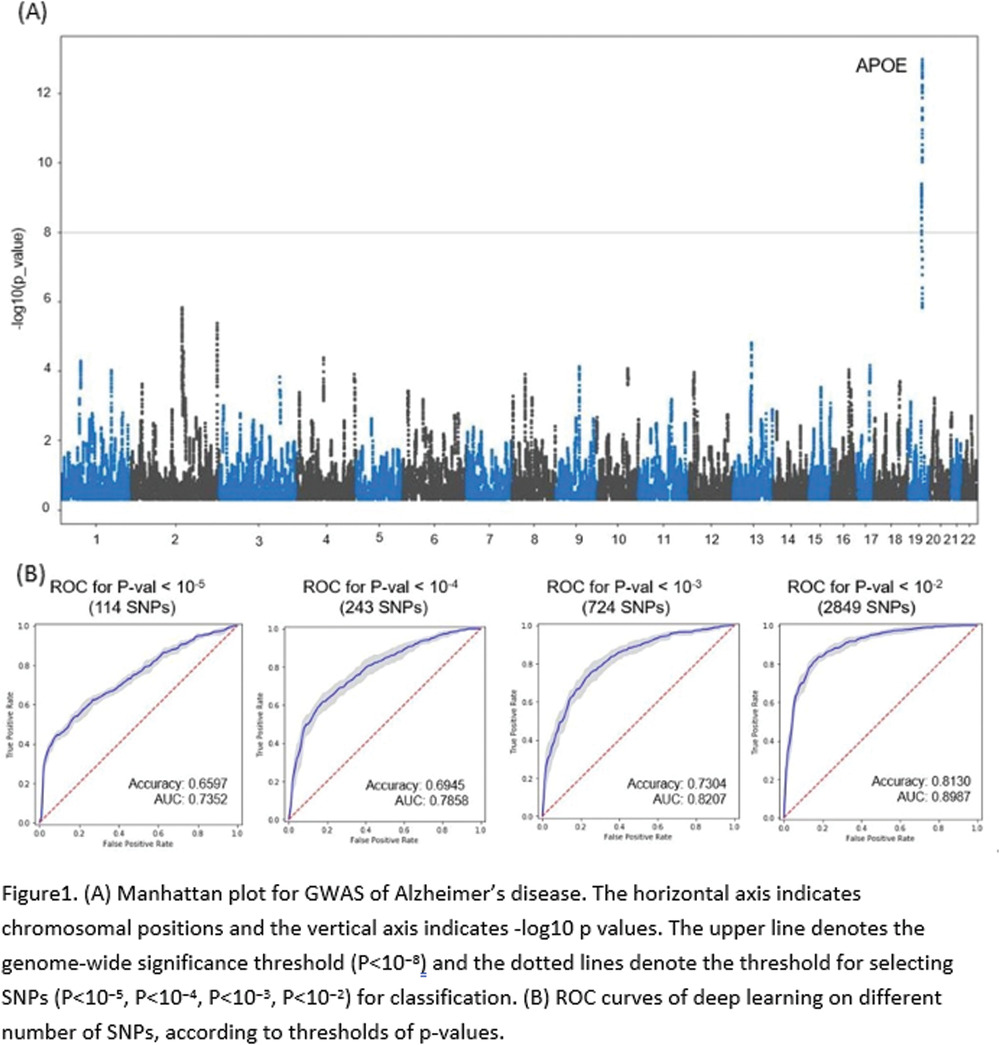
Deep learning–based genome-wide association analysis in Alzheimer’s disease
We used genome-wide genotyping data (12,448,786 SNPs following imputation) from 916 participants in the Alzheimer’s Disease Neuroimaging Initiative (458 cognitively normal controls and 458 AD patie...

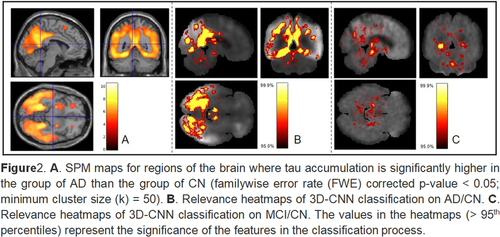
Deep learning detection of informative features in tau PET for Alzheimer’s disease classification
We developed a deep learning-based framework to identify informative features for AD classification using tau positron emission tomography (PET) scans. The 3D convolutional neural network (CNN)-bas...
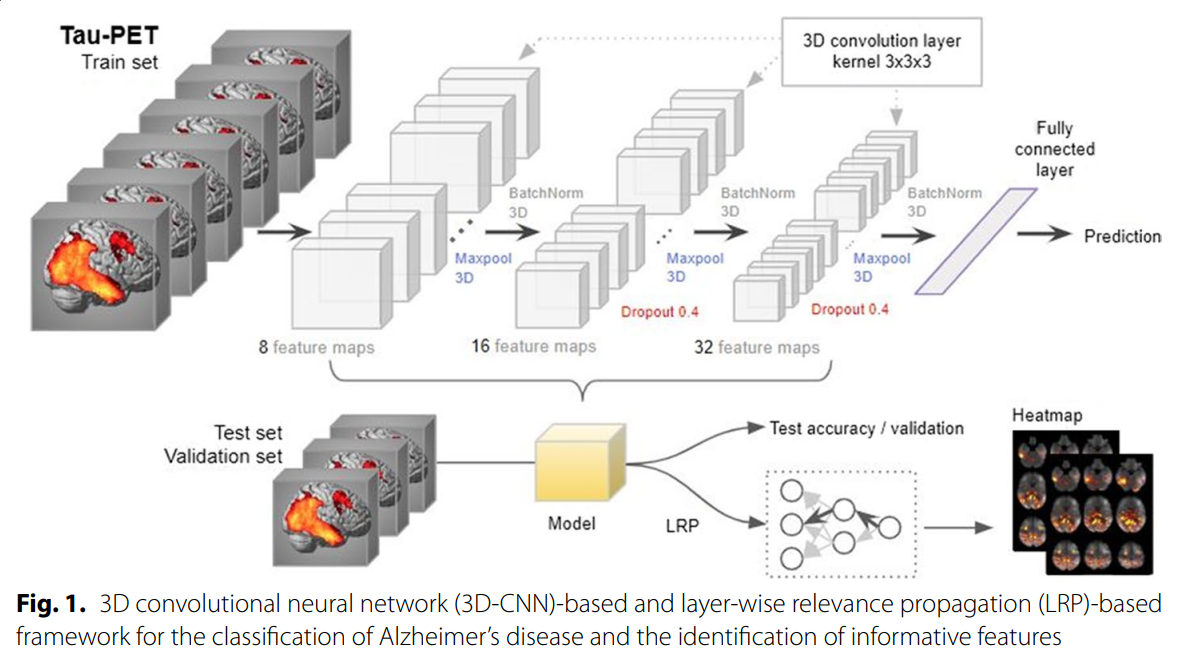
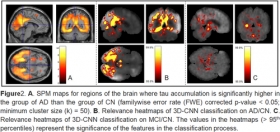
Deep learning detection of informative features in [18F] flortaucipir PET for Alzheimer’s disease classification
We downloaded 458 tau PET images (196 CN, 196 MCI, and 66 AD) from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) and included only one scan per individual. SPM12 was used to process the ta...
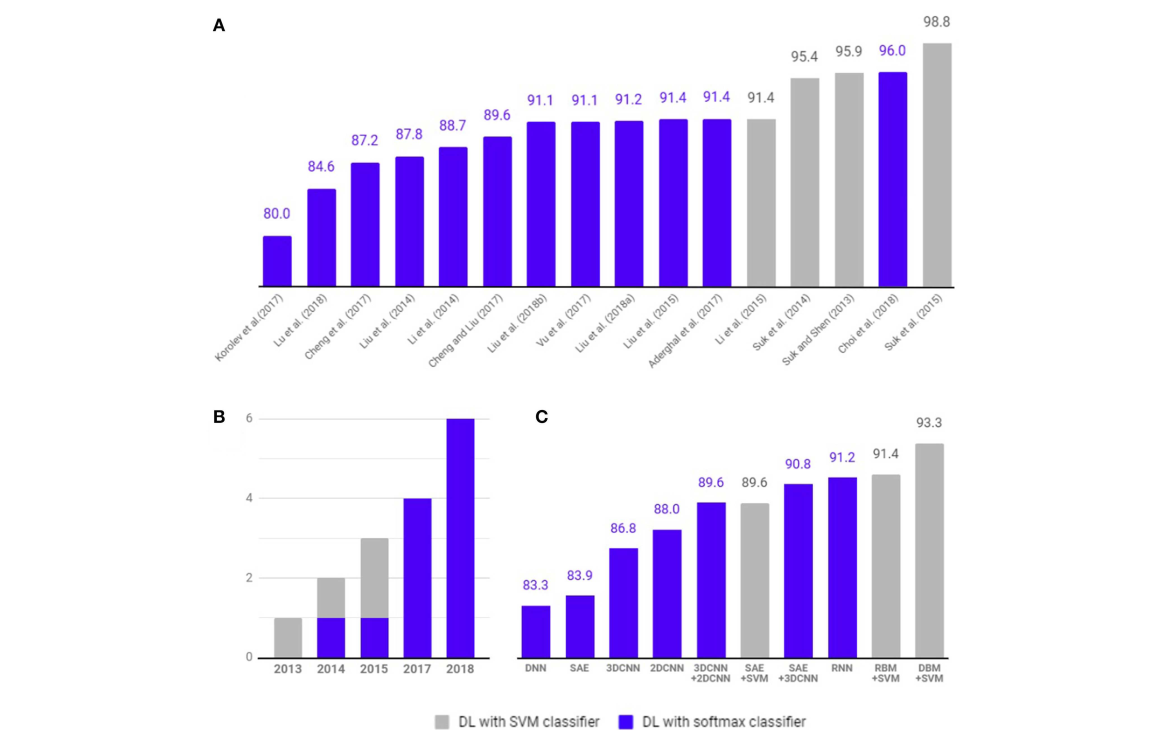
Deep Learning in Alzheimer's Disease: Diagnostic Classification and Prognostic Prediction Using Neuroimaging Data
The application of deep learning to early detection and automated classification of AD has recently gained considerable attention, as rapid progress in neuroimaging techniques has generated large-s...

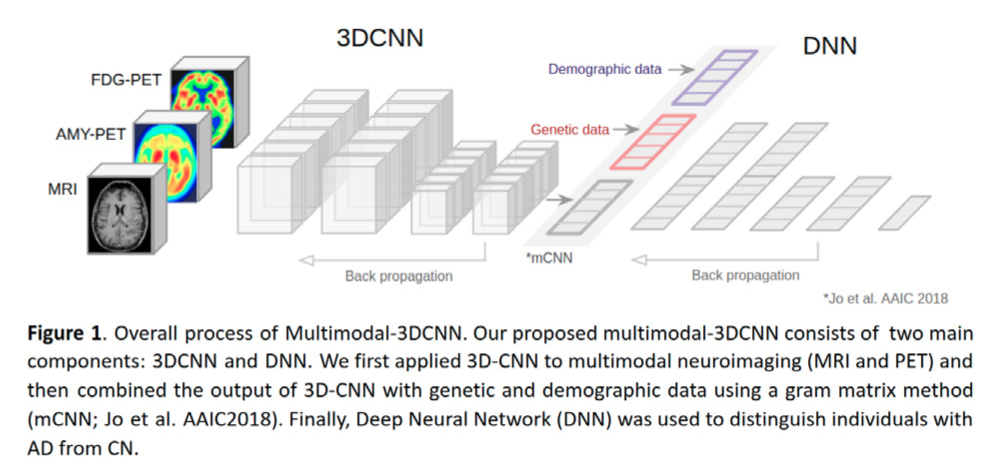
Multimodal-3DCNN: Diagnostic Classification of Alzheimer's Disease Using Deep Learning on Neuroimaging, Genetic, and Demographic Data
Demographic information, 3D MRI and PET image data, and APOE data were downloaded from the ADNI data repository (N=329; 185 CN and 144 AD). In our novel Multimodal-3DCNN approach, we first applied ...
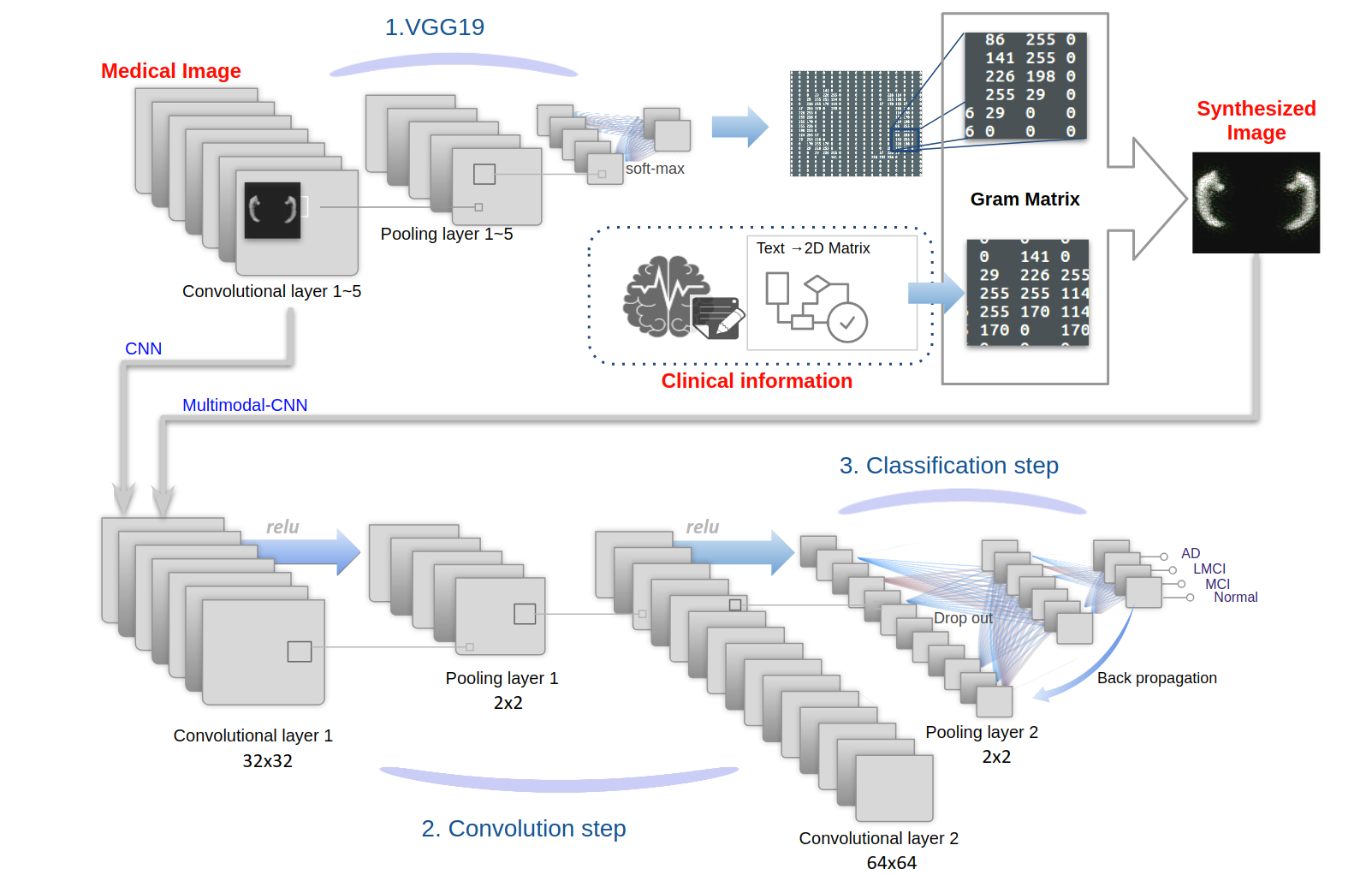
Multimodal-CNN: Improved Accuracy of MRI-based Classification of Alzheimer’s Disease by Incorporating Clinical Data in Deep Learning
Intermediate layers of the CNN were extracted, and the patient's clinical information was added by the gram matrix method. The clinical information was encoded as 2D matrices in this method, and th...
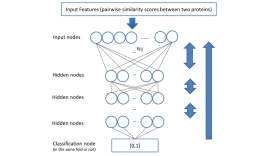
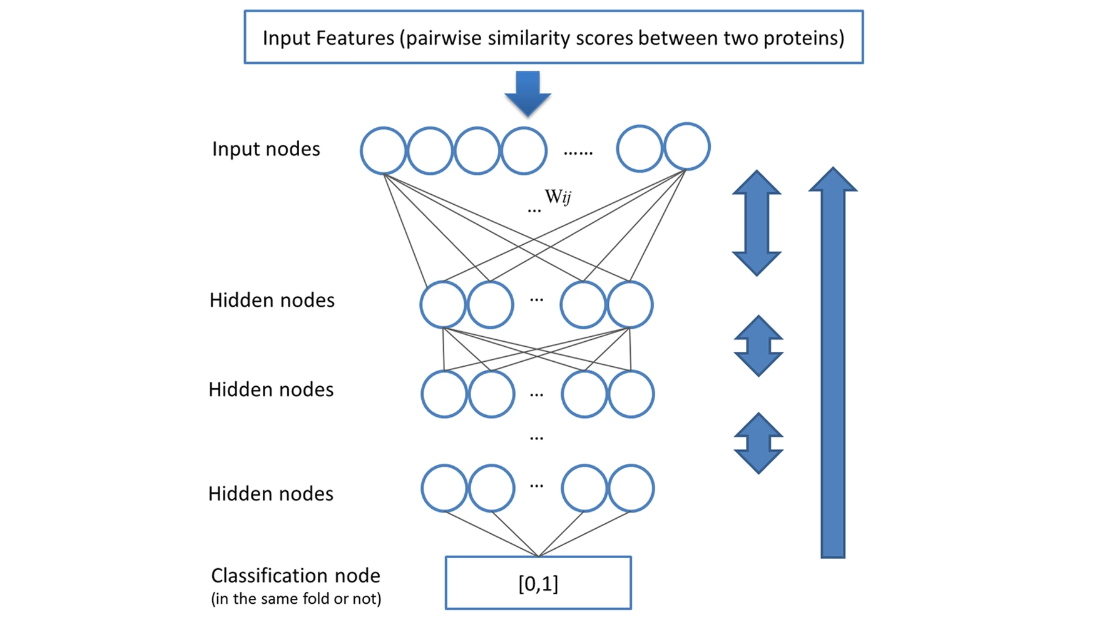
Improving Protein Fold Recognition by Deep Learning Networks
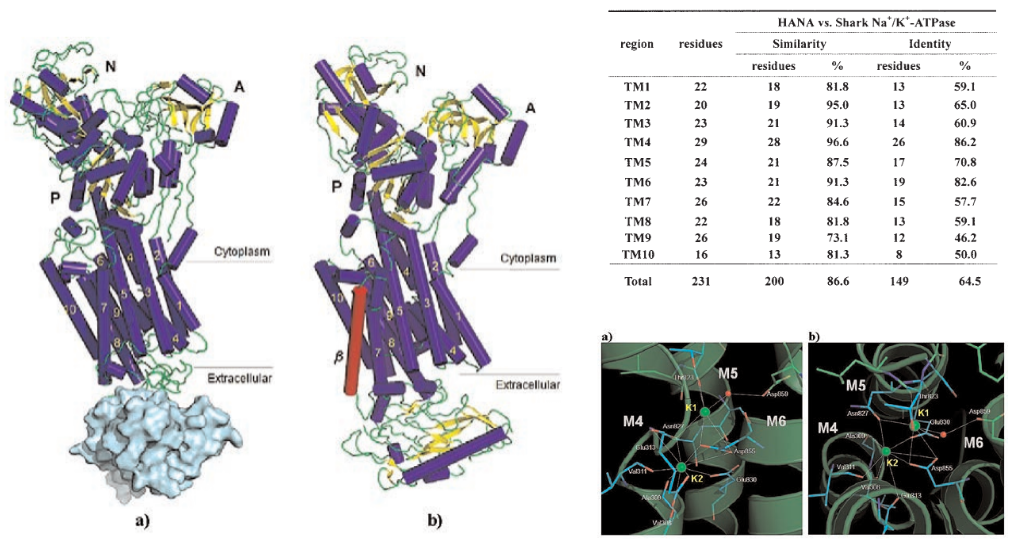
The three–dimensional structure of Heterosigma akashiwo Na+–ATPase (HANA) was predicted by means of homology modeling based on the crystal structure of the K+–bound form of shark Na+/K+–ATPase (PDB...
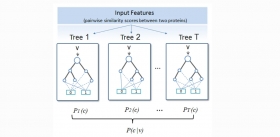
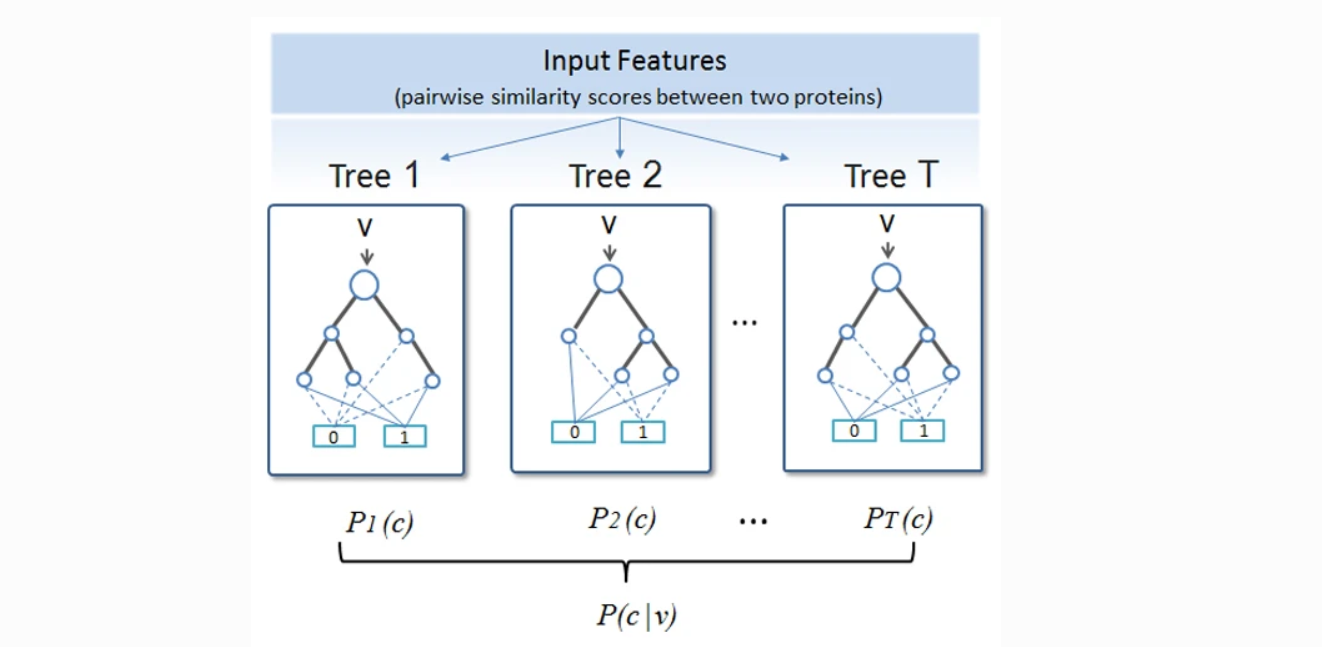
Improving protein fold recognition by random forest
RF-Fold consists of hundreds of decision trees that can be trained efficiently on very large datasets to make accurate predictions on a highly imbalanced dataset. We evaluated RF-Fold on the standa...
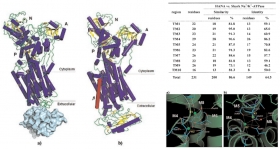
Homology Modeling of an Algal Membrane Protein, Heterosigma Akashiwo Na^+-ATPase
The three–dimensional structure of Heterosigma akashiwo Na+–ATPase (HANA) was predicted by means of homology modeling based on the crystal structure of the K+–bound form of shark Na+/K+–ATPase (PDB...